
Part III of a Blog Series on CQAs & CPPs
In the Cell & Gene Therapy (CGT) field, it is often heard that “the process is the product.” [1, 2] Unlike simple small molecule drugs that are fully characterizable due to their chemically defined formulation, cell products are very complex, and may never be fully characterized. Therefore, especially for CGT, the process is currently an important aspect of defining the end product, along with the Critical Quality Attributes (CQAs). Thus, the FDA has attributed good manufacturing process control as one of the key factors to ensure successful CGT product development. [3] In order to develop a robust manufacturing process, it is important to identify the Critical Process Parameters (CPPs) and define their ranges of acceptable values. In the previous blog, Critical Quality Attributes: Know Their Importance & Limitations in Product & Process Development, we adopted the CPP definition of:
“An input or output process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired product quality.”
Like in any other complex system, the relationship between the input and the output parameters in cell therapy manufacturing is often unknown. We can accept that some CQAs are unknown, but still controllable within certain known “guard rails.” Therefore, to help ensure the maintenance of stable CQAs, we strive to maintain a consistent manufacturing process. This process includes CQAs already defined during product development, as well as the unknown ones. A consistent cell therapy manufacturing process entails a good scientific understanding of the process parameters in each unit operation and implementation of appropriate controls. That starts with identifying the CPPs in manufacturing.
Common Practices in Identifying CPPs
Every manufacturing process has process parameters, and identifying which of the process parameters are “critical” is an important step in any process development program. Several guidelines and examples on identification of CPPs in cell therapy manufacturing have been published [4, 5]. From our experience, it comes down to three steps:
- Perform a gap analysis and identify all process steps.
- Perform studies to quantify the effect of each process parameter on the CQA.
- Define and prioritize CPPs as those parameters that strongly impact CQAs, and establish both a target and acceptable range for each parameter based on experimental data.

It is plainly impossible to evaluate the effect of the hundreds of interconnected parameters that exist within a CGT manufacturing process. Hence, prioritization is important. There are several methods for identifying and prioritizing process parameters, including (i) previous experience in other systems, (ii) existing product/process development experimental data, (iii) published literature, and (iv) quantifiable impacts via a Failure Mode & Effects Analysis (FMEA). A rigorous process development campaign will thoroughly research and investigate to understand which parameters strongly affect the cell CQAs, as well as which are highly variable, and which can be controlled tightly during your cell product manufacturing. As an example of one possible CPP, the temperature of the cell culture inside a bioreactor is usually kept within a tight range as a matter of course, via controlled heating in the equipment. Thus, studying the effect of the temperature on the cell CQAs may warrant low priority. On the other hand, the impact of some different input parameters can prove to be highly variable, such as cell density from different tissue donors or operator handling time. These need to be studied and confined within a range, perhaps with de novo process innovations.
Defining Acceptable Ranges of CPPs
Before we more deeply define acceptable ranges, let’s revisit the concept of input and output parameters. Per Witcher’s definition of CPPs, what does it mean to have your CQAs impacted by both input and output process parameters? In most process steps, the input itself is the parameter, e.g. centrifuge speed. Let’s say that you have done a study and established a range of speeds that works to concentrate your cells with a specified time frame prior to the final fill & finish step, e.g. 300 ×g– 500 ×g for 10-15 minutes. Using any speed/time within this range, your data suggests the process can recover >85% of your cells (which is your established metric), and those cells that you recover after the centrifugation exhibit the same characteristics whether you spin your cells at 300 ×g for 10 minutes or 500 ×g for 15 minutes.
Let’s examine a scenario where an input parameter doesn’t directly affect the CQA. Let’s take a parameter from a formulation step for a media component as an example, where the process parameter of interest is growth factor concentration. For a cellular bioprocess to employ the parameter of 100 µg/L of growth factor, you designed your formulation steps as follows: 1) take 1 vial of growth factor containing 100 µg, 2) dissolve in 10mL of water, and 3) transfer to another container with 990mL of water. Your inputs here are 1 vial (intended weight of 100 µg) and 1L of water. A few things can happen during the formulation that makes your final formulation not exactly that concentration: some of the growth factor powder didn’t get transferred from the vial to the product mixing container, uneven mixing, or simply variability in the vial fill from the manufacturer – these are all the “other” factors that also influence the CPP, which are covered under Critical Material Parameter (CMP) and Critical Equipment Parameter (CEP) described in Part 1 of this blog series.
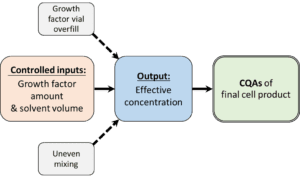
In this particular case, let’s say that all these factors combined can result in a range of concentration from 90 to 110 µg/L. This effective concentration is an output process parameter which cannot easily be controlled without onerous process control measures that may not be appropriate for this parameter. Still, the 10mg/L (-/+) could theoretically have an effect in your product CQA. In some cases, you can monitor the output parameter using ELISAs or in-line sensors, but in other cases, you cannot until you test your final product. Even if this value can be quantified mid-process, it will increase process cost, complexity, and time. That is, decisions need to be made on whether this information is simply to be reported, or if it will trigger an action during the manufacturing (in this case, add more growth factors or dilute with more water). Such actions often increase the manufacturing duration, which by itself can affect your cell product CQAs, as well as increase costs related to labor and GMP suite. To maintain streamlined processes, acceptable ranges for process parameters need to be established to account for these unknown and/or uncontrollable factors. To resolve uncertainty in this case, a development study needs to be done before scaleup, to ensure a growth factor concentration of 90 µg/L and 110 µg/L in your final product will not affect your product performance. If possible, work with your supplier to make sure there are controls around the formulation and fill regarding the growth factor in question.
It’s good practice to choose a target value in the middle of the range that you’ve identified so as to obtain a buffer for the “uncontrollable” output parameters discussed above. Alternatively, the optimum mid-range needs to be ironed out in case there is equipment or operator discrepancy (this, is a whole other topic of IQ/OQ/PQ) – which needs to happen first before any commercial manufacturing. For our purposes here, we assume that all equipment and operators are qualified. In the case of the centrifugation speed and time, you could set your target at 400 ×g for 12 minutes, within the acceptable range of 300-500 ×g and 10-15 minutes. As you gain better understanding and generate more data on your manufacturing process, you can broaden the range until you hit a failure point, i.e. you observe a centrifuge speed that affects the CQAs of the cells post-spin. Broadening the range allows higher process tolerance without changing your manufacturing process.
Process Parameters in MSC Manufacturing
If we apply our 3-step approach (Identify gaps and all process steps -> Quantify effects of parameters on CQA -> Define acceptable parameter ranges) into a standard 3D MSC manufacturing, we will end up with a list of parameters across the various unit operations of the culture process (thaw, inoculation, passage, harvest, formulation & fill) that can impact the MSC product CQAs. For example, during expansion, the CPPs can entail various inputs associated with the MSCs (e.g., cell seeding density), raw material attributes (e.g. media, growth factor concentration), or operational features of the culture vessel (e.g., pH, temperature, dissolved oxygen, and bioreactor agitation). In addition, it is important to keep track of the output parameters throughout culture, such as harvest density and cell viability at passage; cell recovery following harvest and volume reduction; and final cell product Population Doubling Level (PDL). This exercise creates a design space in which the effect of each CPP on the cell CQAs can be quantified, analyzed, and monitored over time. With an appropriate control strategy, both input and output parameters can be kept in their normal operational ranges, which provides the best set of conditions for the production of high-quality MSCs that meet all the known CQAs, and ideally most of the unknown quality attributes as well.
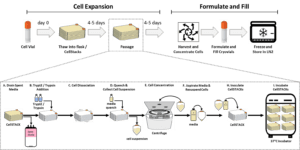
Above, a process overview for MSC expansion and example list of unit operations during passage that are associated with various CPPs, which potentially affect CQAs.
For MSC product development, the ISCT recommends specifying the tissue origin of MSC to highlight tissue-specific properties, since MSCs from different tissue sources have been shown to have different characteristics including proliferative capacity, clonogenic property, immunomodulatory effects, and differentiation capability [6, 7, 8, 9]. Although it’s possible that your standard GMP manufacturing process template may allow room for interchangeable tissue sources so long as product yield is constant, it is expected that different sources of MSCs will result in different CQAs and potencies of your final product, depending on the intended therapeutic application. It is therefore best practice to develop your manufacturing process and the corresponding CPPs around one defined type of MSC source. Based on the heterogeneity and the complexity of MSCs, each MSC product can have unique CQAs—and the corresponding CPPs should be identified case by case.
CPPs Are Important in Maintaining CQAs & Unknown CQAs
Cell therapy product developers usually have a good sense of which final product quality attributes are relevant to their therapeutic application. Yet due to the complex nature of cell therapies, limitations to CQAs do exist, as discussed by Witcher and in our previous blog on Critical Quality Attributes. Oftentimes, analytical assays have limited range or sensitivity to measure shifts to CQAs, thus the term “unmeasurable” CQAs. It is therefore critical to elevate your manufacturing process to the same standards in each run to ensure that there is no shift in these “unmeasurable” CQAs that may ramify into consistency of the downstream product quality.
In conclusion, while this blog lays out the general approaches in identifying CPPs in a manufacturing process, we further expand this paradigm to define CPPs in various MSC production platforms for the next part of this blog series.
At RoosterBio, we offer a wide range of development services which will help you understand the “known unknowns” that affect your CPPs & CQAs.
References
- https://www.biopharma-reporter.com/Article/2017/04/19/For-stem-cells-the-process-is-the-product-says-Pluristem.
- https://www.raps.org/news-and-articles/news-articles/2019/4/establishing-manufacturing-controls-a-hurdle-for.
- Gavin, D.K., Successful Development of Quality Cell and Gene Therapy Products. Food and Drug Administration Center for Biologics Evaluation and Research.
- Campbell, A., et al., Concise Review: Process Development Considerations for Cell Therapy. Stem Cells Transl Med, 2015. 4(10): p. 1155-63. 10.5966/sctm.2014-0294
- Finn, T., Early Stage Manufacturing Considerations for Cell Therapy Products. FDA/CBER: O ffice of Tissues and Advanced Therapies.
- Via, A.G., A. Frizziero, and F. Oliva, Biological properties of mesenchymal Stem Cells from different sources. Muscles Ligaments Tendons J, 2012. 2(3): p. 154-62.
- Hass, R., et al., Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal, 2011. 9: p. 12. 10.1186/1478-811X-9-12
- Kozlowska, U., et al., Similarities and differences between mesenchymal stem/progenitor cells derived from various human tissues. World J Stem Cells, 2019. 11(6): p. 347-374. 10.4252/wjsc.v11.i6.347
- Kwon, A., et al., Tissue-specific Differentiation Potency of Mesenchymal Stromal Cells from Perinatal Tissues. Sci Rep, 2016. 6: p. 23544. 10.1038/srep23544