
“So once you know what the question actually is, you’ll know what the answer means.”
-Douglas Adams, Author of Hitchhiker’s Guide to the Galaxy [1]
From Answers to (Better) Questions…
We all want to find the right drug for the right patient. Yet what if the drug is a cell or a gene? “Precision medicine” and “personalized medicine” are terms that are sometimes used interchangeably, but their meaning is a little different. [2, 3] In the case of cellular therapies involving human mesenchymal stromal/stem cells (hMSCs), the former may be instrumental for the latter to truly flourish. That is, for safe and effective treatments to dovetail with each individual patient’s need (as per “personalized”), a more rigorous understanding of both the disease process and patient cohort “omics” will be necessary (as per “precision”). As we’ll see in this blog, allogeneic-administered hMSCs comprise a biomaterial that can invigorate both the nearer-term “precision” and longer-term “personalized” objectives for cell and gene therapies (CGTs).
The US National Research Council (NRC) published a policy document in 2011, [4] “Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease.” In it was a call to action to “Integrate Data to Construct a Disease Knowledge Network” as well as share data, incentivize public-private partnerships, crowd-source complex problems, and protect individual privacy rights. In other words, the vast data from newly-discovered pharmacogenomic disease biomarkers could conduit with patients directly at point of care, informing better optimized treatments and treatment combinations. Yet, this language seems to be a semantic shift from the rarefied ideal of “personalized medicine,” which could imply that each patient gets an individualized treatment. With apologies to Douglas Adams, [1] the gaps in our understanding might require us to first backtrack from seeking the “Ultimate Answer” to therapeutics, and instead opt for the pragmatism of precision medicine. Perhaps we need to first get a little empirical, to investigate patterns on the “Galaxy” of interacting disease phenomena (“answers that Guide to more meaningful questions”). Thereafter, we can test these educated guesses with corresponding statistical and methodological rigor (“answers via deduction”). Practically speaking, such iterative loops of induction and deduction need groups of patients, not single individuals.
Fig 1. (Below) Precision medicine is now a more widely used term in both biomedical research (left), as well as in research related to cell therapy (right).
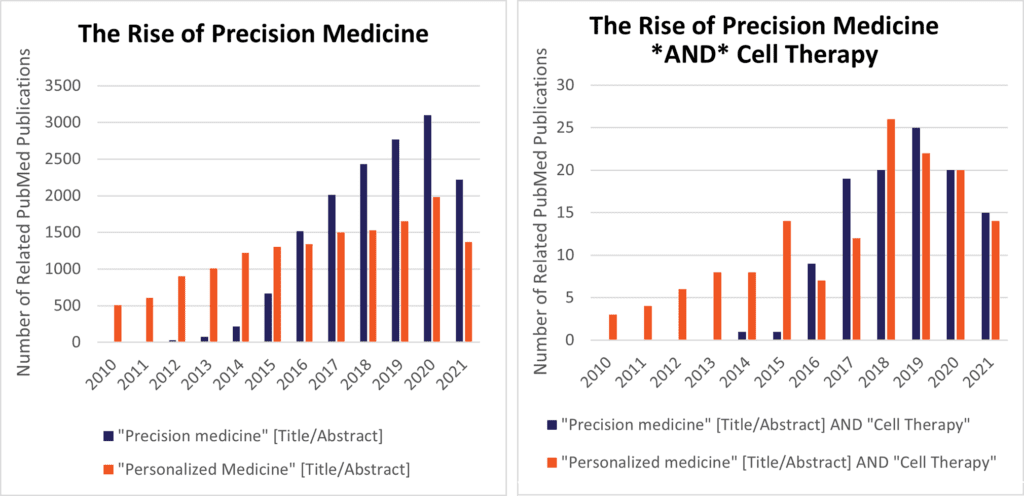
Precision medicine (PrM) as a concept mushroomed after President Obama answered the NRC’s 2011 call to action in his 2015 State-of-the-Union speech (see Fig. 1), launching the National Precision Medicine Initiative, which infused $215M into the NIH’s fy2016 budget. Conventional questions for PrM might involve how the pharmacogenomics of cytochrome P450 affect different patients’ reactions to cytotoxic cancer drugs or opioids, [5] or polymorphisms of Fc-gamma receptors and their relationship to mAb therapeutics or autoimmune disease. [6] Yet, where does that leave “next generation” approaches like hMSC-based cell therapy? CGTs are necessarily more complex to test, manufacture, and administer, hence a challenge. However, cells are also much easier than small molecule pills to be “programmable” with genetic or media factors upstream in their preparation, hence a great opportunity. To zero in on this opportunity, two complementary approaches can help bridge the gap between PrM principles and cellular therapies:
- Tailor the grouping of patient study populations (according to molecular phenotypes) to pair with one specific cell therapy product; or
- Tailor the cell therapy product to optimally pair with a specific patient grouping
We’ll briefly explore some examples of each theme in this blog.
Find the “Right” Patients for Your hMSCs
Allogeneic cell therapies are a forte of hMSCs and related cell types. Due to capacity of these cells for extended population doubling in cGMP bioreactor cultures, tens to 1000s of doses (~100M-1000M cells) can be grown out from a single qualified donor of umbilical cord, bone marrow aspirate, or adipose tissue. Because hMSCs exhibit very low immunogenicity or even promote tolerance, one healthy donor can be suitable as a common starting material for safe administration into non-tissue matched patients. Unlike allo-transplants of T-cells for CAR-T therapy or allo-HSCs for hematopoietic stem cell transplantation, allo-hMSCs pose very low risk of acute rejection reactions—and in fact have been under review as agents to combat GvHD in Phase III trials. With allogeneic hMSCs, one also doesn’t have to wait for a long period of ex vivo expansion after extraction prior to re-transplantation, as per autologous hMSCs. Nevertheless, the penultimate hMSC clinical efficacy success story is yet to be written, with only a handful of approved products approved outside of the USA, and none yet by the US FDA. It stands to reason that with a track record of over 20-years of clinical development, future hMSC therapeutics can only benefit from more adept trial design, based on relevant parameters and endpoints, informed by panels of disease biomarkers.
Although a growing list of hMSC products did not cross the finish line out of late-phase trials, patient histories and diagnostic samples create opportunities for retrospective analyses using newer analytical toolsets via -omics and advanced computational processing. In one early-phase trial example, the group of Dr. Joshua Hare and colleagues with the University of Miami examined genotyping from a 105-gene panel via 37 responders and non-responders to the MSC therapy in their POSEIDON-DCM trial (NCT01392625) for non-ischemic dilated cardiomyopathy (NIDCM). [7] Patients were classified as having either (i), “benign” sets of gene sequences; (ii), variants of unknown significance or “VUS;” or (iii), patients with known pathogenic gene variants. Here, patients with no VUS or no pathogenic gene variants (V-minus) showed significantly better endothelial cell activity and quality of life (MLHFQ scores). V-minus patients and VUS patients significantly improved their ejection fraction (%EF) at 12 months, whereas pathogenic variant (PV+) patients appeared to decline in function. In addition, at the final follow-up, all six V-minus patients were still alive, whereas the VUS group of 20 reported 85% survival, and only 40% remained alive in the PV+ group. Although this study’s patient sample size was small, one possible “take home message” could signify that patients with no VUS or pathogenic gene polymorphisms might positively respond to hMSC-based cell therapies and forego LVAD treatment, while patients with pathogenic variants might need an accelerated path to an LVAD or a heart transplant. Follow-on trial designs that use hMSCs could thus target a more stratified patient population at risk of heart failure, and further reduce the risk of landing short of the endpoint in later phase trials.
In addition to patient genotypic differences, several have noted that the microenvironment of re-implanted hMSCs could exert profound effects on their behavior and, possibly, patient outcome. [8, 9] Consequently, differences in the injection route (e.g., IV vs. local) would affect hMSC exposures as they pass through different microenvironments in vivo. For example, variable concentrations of local oxygen, IL-1, TNF-a, and SDF-1a and may affect hMSC differentiation, immune modulation activity, proliferation, and chemotaxis. Conversely, related antagonist pharmacologic interventions in patients like hyperbaric therapy, corticosteroids, Kineret®, Enbrel®, and Mozobil® could also affect outcomes of hMSC therapy. If these and other variable factors are not taken into consideration, very heterogeneous bioactivity could result from superficially similar groups of patients, affecting clinical study p-values.
A related insight into the microenvironment’s metabolome and proteome may emerge via studies of hMSCs used for acute respiratory distress syndrome (ARDS). In mice—and also large in vivo animal and human ex vivo lung models of ARDS—hMSCs related to Dr. Michael Matthay’s preclinical initiatives repeatedly yielded dramatic and rapid improvements in infection-damaged organs. Much to the astonishment of many, the phase IIa START trial for hMSCs in human patient ARDS failed to show benefit, and 28-day mortality was twofold higher (non-significant) in the MSC group. [10] Accordingly, Islam et al (2019) sought to dissect canonical biomarkers (TAC, ~ oxidative stress; IL-6 ~ NF-kB related inflammation; FN ~ fibrosis) in lung microenvironment that could serve as guides for anti-ARDS hMSC therapy. Animals in this study fared worse with the MSCs when injured with stimuli that increased bronchoalveolar levels of IL-6 and fibronectin. [11] Most intriguingly, these MSCs improved outcomes when tweaked with genetic engineering to express anti-inflammatory IL-10 or anti-fibrotic HGF. One possible hypothesis to explore for further study is that mechanical lung injury by ventilator could be benefitted by hMSCs, yet hMSCs used to improve infection-related ARDS (e.g., COVID-19) would need further genetic modification and/or co-treatments. Nevertheless, in Dr. Matthay’s reply to this study, [12] it was noted that IL-6 plasma levels were collected via the START trial patients, but these didn’t correlate with patient outcome. (Fibronectin or TAC readouts weren’t assessed.) These puzzling results (and some later ones related to COVID-19 [13]) suggest that more studies may be needed to help resolve these many complex variables in play. We [14] and others [15, 16] have suggested that MSCs genetically armed to combat a vicious cycle of NETosis and coagulopathy may be worth further investigation.
Find the Right hMSCs for Your Patients
What’s more of a precise “fit” for a cell therapy source than a patient’s own tissue? It’s well-known that hMSCs can be safely administered from autologous sources—particularly adipose tissue (AT)—where large cell volumes are rapidly available from a routine lipoplasty. Adipose tissue contains 100–500 fold more AT-MSCs and/or adipose tissue stem cells (ASCs) compared to bone marrow. [17] However, quality of AT-MSCs depends on extraction methods and the individual patient sample. ASCs/AT-MSCs are reportedly a heterogeneous mix, where the panel of CD- markers does overlap with ISCT hMSC criteria [18, 19] but also can reflect various stromal cells, endothelial progenitors, pericytes, and hematopoietic cells as well as MSCs. Stating the obvious, different cell isolation methods from different patients, of different ages, and expanded with even slightly different culture conditions can and do yield different results in the clinic—a complex set of variables to quality control. On the other hand, a therapeutic “force multiplier” for autologous MSCs might be a genetic modification, such that the major balance of pharmacologic potency is delivered by the artificial gene—and less, by the cell. As we noted in a previous blog, [20] different sources of hMSCs can have different “talents.” Generally, the trait of extended population doubling is much superior in hMSCs via umbilical cord (UC-) than from AT-MSCs. Thus, genetic enhancement to drive therapeutic AT-MSCs could be more of a challenge than UC-MSCs, if the transduction/transfection method is low efficiency. With possibly heterogeneous cell materials expanded out from patient to patient, similar hiccups once observed for many autologous CAR-Ts could also apply to gene-engineered, autologous AT-MSCs. That said, autologous sourced AT-MSCs will remain highly promising to source “personalized” medicines for the foreseeable future, all while new and meaningful “precision medicine” insights are continuously gleaned from them.
Tools to “fingerprint” micro-RNA (miRNA) profiles via hMSCs or their secretomes remain tantalizing to evaluate hMSC therapeutics by scanning for their most enriched and most related biological process signatures. [21, 22] For example, Ragni, et al (2019) discovered key miRNA reference genes from profiling the extracellular vesicle (EV) “miRome” secreted via AT-MSCs that were subjected to immune cytokine priming. EVs from MSC donors that were best “licensed” by the pre-treatment most abundantly expressed the reference miRNA in secreted EVs. These authors noted that similar methods could be readily translatable to an allogeneic human therapeutic for osteoarthritis, where less potent donors of cell manufacturing starting materials could be excluded. [21] Since EVs with miRNA cargo are abundantly secreted from MSCs,[23, 24, 25] it’s no surprise that in-line-sampled, real-time measurements of miRNA-EV prevalence across different biomanufacturing platforms can reveal insights of unprecedented sophistication toward optimal quality-controlled culture conditions.[26]
Availability of the fully sequenced human genome and epi-genome could map out a multi-omics dashboard for hMSCs under bioreactor culture conditions, just like their CHO cell predecessors.[27] That is, the dynamic feedback between external metabolite concentration, the cells’ metabolic flux, and CRISPR-targetable bioenergetics (or senescence) pathways could be monitored, reported on, and manipulated rapidly to ensure the highest-quality cell products for a particular disease indication. Moreover, not only can toolkits such as RNA-seq, chromatin accessibility assays, and their surrogate biomarkers distinguish between hMSC tissue origin[28] or differentiation fate,[29, 30] they can also help establish universal reference materials and baseline standards between formerly disparate MSC material batches and processes.[31]
The creative possibilities to employ hMSCs as carriers of therapeutic genes (or release EVs that bear gene-modifying cargoes in vivo or in ex vivo production) are nearly inexhaustible. These engineered gene circuits allow highly precise disease targeting down to the specificity of single base pair with technologies such as CRISPR. Presently, we are aware of one posted clinical trial from University Hospital, Montpellier (NCT03855631), where 4 of 8 Kabuki Syndrome patients are to be treated with MSCs that have a transcriptionally-targeted CRISPR system to increase the expression of the wild KMT2D allele to restore the functional activity of MLL4. In another demonstration of CRISPR-Cas for a model of Parkinson’s disease (PD) in rats, the CRISPR is used to target a therapeutic expression cassette into a genomic safe harbor in MSCs; this prospective cell-gene therapy specifically ameliorates Parkinson’s by targeting albumin that is lesioned with advanced glycation end products (AGEs) via a secreted, soluble receptor for AGEs [32]. PD may present in complex ways with differing underlying subtypes [33]—according to known and unknown gene polymorphisms—and hence might be an attractive disease target via precision medicine therapies that use hMSCs as the “chassis” for these kinds of gene circuits.
In well-known examples via non-MSC bioproduction cells, siRNA-cargoed and RVG-peptide-targeted EVs/exosomes were shown to ameliorate mouse models of Alzheimer’s Disease [34] and opiate addiction. [35] In another example that does use hMSCs as producers, Dr. Kalluri’s group from MD Anderson Cancer Center aims to specifically target kRAS (G12D mutant) driven pancreatic cancer with EVs (“iExosomes”) engineered for longer half-life and delivery of a packaged siRNA. [36] A wide range of additional pioneering concepts for engineered EVs (including EVs engineered in hMSCs) are explored in an excellent review, featuring authors Oscar Wiklander, Meadhbh Brennan, Jan Lötvall, Xandra Breakefield, and Samir EL Andaloussi. [37] Of uniquely high impact for precision EV therapeutics might be an adaptation of the Kyoto University’s “NanoMEDIC” approach to bioproduction via MSC-EVs—this molecular tech directs the CRISPR proteins and gRNA ribonuncleoparticles (RNPs) into EVs with extremely high efficiency. Then these EVs internalize into cells in a targeted manner–also with very high efficiency. NanoMEDIC appears to work against in vivo mouse models of Duchenne muscular dystrophy (DMD) by “surgically” excising defective dystrophin exons. [38] Like PD, DMD also presents itself as an intriguing target for precision medicine, since there are many varying polymorphisms and subtypes across its 2.2 megabases and 79 exons of genomic sequence.
From Better Questions to Even Better Answers
In this blog, we’ve explored possible examples of big data sets (e.g., the Genome of ~3,000,000,000 base pairs) to direct the best MSC cellular starting materials (or MSC-EVs) toward the “ideal” patients. And, conversely, we looked at possible ways of directing the “best” patients toward the ideal kinds of MSCs. Either way, to accomplish both goals, it will be helpful to call upon non-biased, sophisticated computational, and high throughput -omics data sets to interrogate the astonishingly complex biological networks that go awry in disease. Simply put, we need a better understanding of where human homeostasis is likely to be fragile, and where it’s robust at a molecular network level. True, this is a more difficult endeavor with complex living cells (like hMSCs) than with small molecule pills. Yet, precision medicine for hMSCs remains necessary not in spite of, but because of, the complexity of cells vs. small molecule pharmaceuticals.
Take heart, though! With apologies (again) to Douglas Adams, we don’t need a full “Encyclopedia Galactica” of bioinformatics to get moving, only enough knowledge for something like a “Hitchhiker’s Guide” emblazoned with the friendly letters, “DON’T PANIC.” If we have just enough to formulate a meaningful question based on clear observations, we’ll surely be soon spiraling in progressively bolder permutations toward the precise therapeutic answers that will benefit patients in need—and at increasingly lower costs. Isn’t that what precision medicine is about?
To help make precision medicine work for you in cellular or gene therapy, RoosterBio has engineered a growing product system of high-performance media ancillary materials and hMSC cellular starting materials that perform consistently through R&D to cGMP and clinical production scales. Since opening our doors in 2014, we’ve worked with over 300 customers to assist in their academic and clinical translational needs, who have published hundreds of peer-reviewed journal articles that cite our materials’ use in their methods. Why not check out the Publications section of our Knowledge Center to learn how these investigators are identifying compelling questions and unexpected answers through use of our products and services toolkits?
References
- Adams, Douglas, The Hitchhiker’s Guide to the Galaxy. 1979: Pan Books.
- Bresnick, Jennifer, What Are Precision Medicine and Personalized Medicine? 2018: healthitanalytics.com.
- Khoury, Muin, in The Shift From Personalized Medicine to Precision Medicine and Precision Public Health: Words Matter! 2016, blogs.cdc.gov: Centers for Disease Control and Prevention (CDC).
- in Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. 2011: Washington (DC).
- Preissner, S. C., et al., Polymorphic cytochrome P450 enzymes (CYPs) and their role in personalized therapy. PLoS One, 2013. 8(12): p. e82562. 10.1371/journal.pone.0082562
- Nagelkerke, S. Q., et al., Genetic Variation in Low-To-Medium-Affinity Fcgamma Receptors: Functional Consequences, Disease Associations, and Opportunities for Personalized Medicine. Front Immunol, 2019. 10: p. 2237. 10.3389/fimmu.2019.02237
- Rieger, A. C., et al., Genetic determinants of responsiveness to mesenchymal stem cell injections in non-ischemic dilated cardiomyopathy. EBioMedicine, 2019. 48: p. 377-385. 10.1016/j.ebiom.2019.09.043
- Greco, S. J. and P. Rameshwar, Microenvironmental considerations in the application of human mesenchymal stem cells in regenerative therapies. Biologics, 2008. 2(4): p. 699-705. 10.2147/btt.s2765
- Patel, S. A., et al., Personalizing Stem Cell Research and Therapy: The Arduous Road Ahead or Missed Opportunity? Curr Pharmacogenomics Person Med, 2010. 8(1): p. 25-36. 10.2174/1875692111008010025
- Matthay, M. A., et al., Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): a randomised phase 2a safety trial. Lancet Respir Med, 2019. 7(2): p. 154-162. 10.1016/S2213-2600(18)30418-1
- Islam, D., et al., Identification and Modulation of Microenvironment Is Crucial for Effective Mesenchymal Stromal Cell Therapy in Acute Lung Injury. Am J Respir Crit Care Med, 2019. 199(10): p. 1214-1224. 10.1164/rccm.201802-0356OC
- Matthay, M. A., et al., Precision medicine for cell therapy in acute respiratory distress syndrome – Authors’ reply. Lancet Respir Med, 2019. 7(4): p. e14. 10.1016/S2213-2600(19)30083-9
- Keown, Alex, Mesoblast Shares Plunge After Stem Cell Therapy Fails COVID-19 Study. 2020: BioSpace.
- Carson, Jonathan, When the NETs Fail in COVID-19’s Pulmonary Fire: hMSCs to the Rescue, in RoosterBio Blog. 2020: RoosterBio.
- Wang, J., et al., Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol, 2020. 108(1): p. 17-41. 10.1002/JLB.3COVR0520-272R
- Khoury, M., et al., The Coronavirus Pandemic: A Pitfall or a Fast Track for Validating Cell Therapy Products? Stem Cells Dev, 2021. 30(3): p. 119-127. 10.1089/scd.2020.0122
- Facklam, A. L., L. R. Volpatti, and D. G. Anderson, Biomaterials for Personalized Cell Therapy. Adv Mater, 2020. 32(13): p. e1902005. 10.1002/adma.201902005
- Horwitz, E. M., et al., Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy, 2005. 7(5): p. 393-5. 10.1080/14653240500319234
- Viswanathan, S., et al., Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT(R)) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy, 2019. 21(10): p. 1019-1024. 10.1016/j.jcyt.2019.08.002
- Carson, Jonathan, hUC-MSCs Are Unique: Are the Biomaterials, Bioprocesses, & Clinical Product Developers Now “in Accord”? 2021: RoosterBio, Blog.
- Ragni, E., et al., Insights into Inflammatory Priming of Adipose-Derived Mesenchymal Stem Cells: Validation of Extracellular Vesicles-Embedded miRNA Reference Genes as A Crucial Step for Donor Selection. Cells, 2019. 8(4). 10.3390/cells8040369
- Ferguson, S. W., et al., The microRNA regulatory landscape of MSC-derived exosomes: a systems view. Sci Rep, 2018. 8(1): p. 1419. 10.1038/s41598-018-19581-x
- Carson, Jonathan. It’s Not Rocket Science – “MSC-EVs” Have the Right Stuff. 2021; Available from: https://bit.ly/3B7Byn8.
- Yeo, R. W., et al., Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev, 2013. 65(3): p. 336-41. 10.1016/j.addr.2012.07.001
- Haraszti, R. A., et al., Exosomes Produced from 3D Cultures of MSCs by Tangential Flow Filtration Show Higher Yield and Improved Activity. Mol Ther, 2018. 26(12): p. 2838-2847. 10.1016/j.ymthe.2018.09.015
- Lam, A. T., et al., Multiomics analyses of cytokines, genes, miRNA, and regulatory networks in human mesenchymal stem cells expanded in stirred microcarrier-spinner cultures. Stem Cell Res, 2021. 53: p. 102272. 10.1016/j.scr.2021.102272
- Lewis, N. E., et al., Genomic landscapes of Chinese hamster ovary cell lines as revealed by the Cricetulus griseus draft genome. Nat Biotechnol, 2013. 31(8): p. 759-65. 10.1038/nbt.2624
- Ho, Y. T., et al., Chromatin accessibility identifies diversity in mesenchymal stem cells from different tissue origins. Sci Rep, 2018. 8(1): p. 17765. 10.1038/s41598-018-36057-0
- Wang, C., et al., RNA-Seq Based Transcriptome Analysis of Endothelial Differentiation of Bone Marrow Mesenchymal Stem Cells. Eur J Vasc Endovasc Surg, 2020. 59(5): p. 834-842. 10.1016/j.ejvs.2019.11.003
- Khan, A. A., et al., Significant transcriptomic changes are associated with differentiation of bone marrow-derived mesenchymal stem cells into neural progenitor-like cells in the presence of bFGF and EGF. Cell Biosci, 2020. 10: p. 126. 10.1186/s13578-020-00487-z
- Robb, K. P., et al., Mesenchymal stromal cell therapy: progress in manufacturing and assessments of potency. Cytotherapy, 2019. 21(3): p. 289-306. 10.1016/j.jcyt.2018.10.014
- Lee, J., et al., CRISPR/Cas9 Edited sRAGE-MSCs Protect Neuronal Death in Parkinsons Disease Model. Int J Stem Cells, 2019. 12(1): p. 114-124. 10.15283/ijsc18110
- Bloem, B. R., M. S. Okun, and C. Klein, Parkinson’s disease. Lancet, 2021. 397(10291): p. 2284-2303. 10.1016/S0140-6736(21)00218-X
- Alvarez-Erviti, L., et al., Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol, 2011. 29(4): p. 341-5. 10.1038/nbt.1807
- Liu, Y., et al., Targeted exosome-mediated delivery of opioid receptor Mu siRNA for the treatment of morphine relapse. Sci Rep, 2015. 5: p. 17543. 10.1038/srep17543
- Kamerkar, S., et al., Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature, 2017. 546(7659): p. 498-503. 10.1038/nature22341
- Wiklander, O. P. B., et al., Advances in therapeutic applications of extracellular vesicles. Sci Transl Med, 2019. 11(492). 10.1126/scitranslmed.aav8521
- Gee, P., et al., Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat Commun, 2020. 11(1): p. 1334. 10.1038/s41467-020-14957-y