
Harnessing “Infinite Diversity in Infinite Combinations” via Synthetic Biology for Mesenchymal Stem Cells & Their Exosomes/Extracellular Vesicles as Candidate Leads
In Star Trek lore, there’s a mantra that uncannily reflects to synthetic biology: “Infinite Diversity in Infinite Combinations.” IDIC, the basis of Vulcan philosophy, celebrates not just the vast array of variables in the universe, but also the huge complexity springing from even a small array of testable components when optimizing a biological therapeutic. How does this apply to you? The simple answer is that in complex, open biological systems, context matters. You cannot always predict how interchangeable molecular parts “play well” with each other because these parts may be subtly interconnected. Emergent phenomena can mysteriously appear when new sequence configurations operatively link up—the whole isn’t always the sum of its parts. Before you “think big” and scale up your CMC bioprocess, consider how it could benefit your team to first “scale out” your candidate discovery and optimization with therapeutics like synthetic exosomes and/or extracellular vesicles (EVs). To pan for the golden engineered extracellular vesicle therapeutic, harness the power of IDIC!
Not to belabor this too long, but here’s an analogy. Imagine a souped-up keyboard (~ a cell), loaded with soundfonts for every imaginable digital waveform and any frequency (~ the genome). You plunk on the notes C, E, and F (~ transcripts) together. Out hums a lovely and sweet C-major… Sunlight shines warmly on your shoulder and the Rooster heralds a bright morning. Then you bang on C, Eb, and F; the only difference is that your middle finger moves ever slightly, down a semitone to the black key. The sound is now a melancholy C-minor…A thick miasma rolls in; a murder of crows gathers menacingly. Gene expression or cell signaling is like that. It doesn’t matter if genes “C” and “F” are still active when the switch between genes “E” and “Eb” (in the context of C/F) proves decisive for a cell’s fate. So, when you endeavor to employ the cell as an “instrument” for exosome/extracellular vesicle manufacture, perhaps it might be wise to conjure that special ambience by empirically surveying and cataloging a broad repertoire of “good chords” to try?
Exosomes/extracellular vesicles are now approached as an attractive technology platform by pipeline strategists [1, 2] because they can be targetable like monoclonal antibodies (mAbs), [3, 4, 5, 6, 7] and yet also encapsulate therapeutic activities as per liposomes or LNPs. [6, 7, 8, 9, 10, 11, 12, 13] Secreted from animal and human cells, they can be IV-injected without eliciting an acute allergic response, and in certain contexts may even induce an immune cooling effect in vivo. [14] There are even various putative demonstrations wherein these small, lipid-bound, cell-derived particles can encapsulate viral gene therapy vectors like AAV, and, in turn, enhance in vivo uptake and cloak them from attack by neutralizing antibodies (nAbs). [15, 16, 17, 18] If practicable, such technology could open gene therapies to much larger cohorts of vector-seropositive individuals. Most importantly, exosomes can be engineerable with segmented, modular DNA or RNA “software”—ported into cellular “hardware” to customize exosome function toward novel clinical products with improved efficacy and manufacturability.
A recently accelerated pace of exosome-related clinical trials, journal publications, and patent applications confirms exuberance for this topic of investigation. The earliest clinical activity for exosomes as therapeutics has mostly comprised unmodified biological materials, a large majority of which are harvested from conditioned media via mesenchymal stromal/stem cells (hMSCs). This is chiefly because of reports to suggest that hMSC secretomes and/or derivative isolated exosomes are dominant contributors to these cells’ therapeutic effect. [19, 20]
However, there exist contrasting reasons to tranquilly embrace non-engineered, “vanilla” exosomes and eschew the force multiplier of synthetic biology. One particular reason is not trivial—genetically engineering exosomes could create a challenging technical barrier to entry atop an already complex drug platform. How to surmount the obstacle of genetic material that (potentially) opens a whole extra dimension of drug product and process monitoring for the CMC team? Although some may answer this objection in different ways, many extracellular vesicle product developers recognize a downstream tradeoff that heavily weighs in their favor. That is, when there is an optimized fit between the novel molecular (DNA or RNA) and cellular starting material, the drug product will prove exceptional enough to outweigh the time and trouble to design, build, and test it. This rationale is not unprecedented. It has been historically applied to heavily engineered CHO cells and their recombinant monoclonal antibody (mAB) products—a platform that now accounts for over $200B in annual global sales.
In Figure 1 (below), we illustrate one of many possible example schemes to mitigate future bioproduction hiccups by selection of a robust handful of engineered exosome/extracellular vesicle candidates from early, back-end discovery-phase HTS panning.
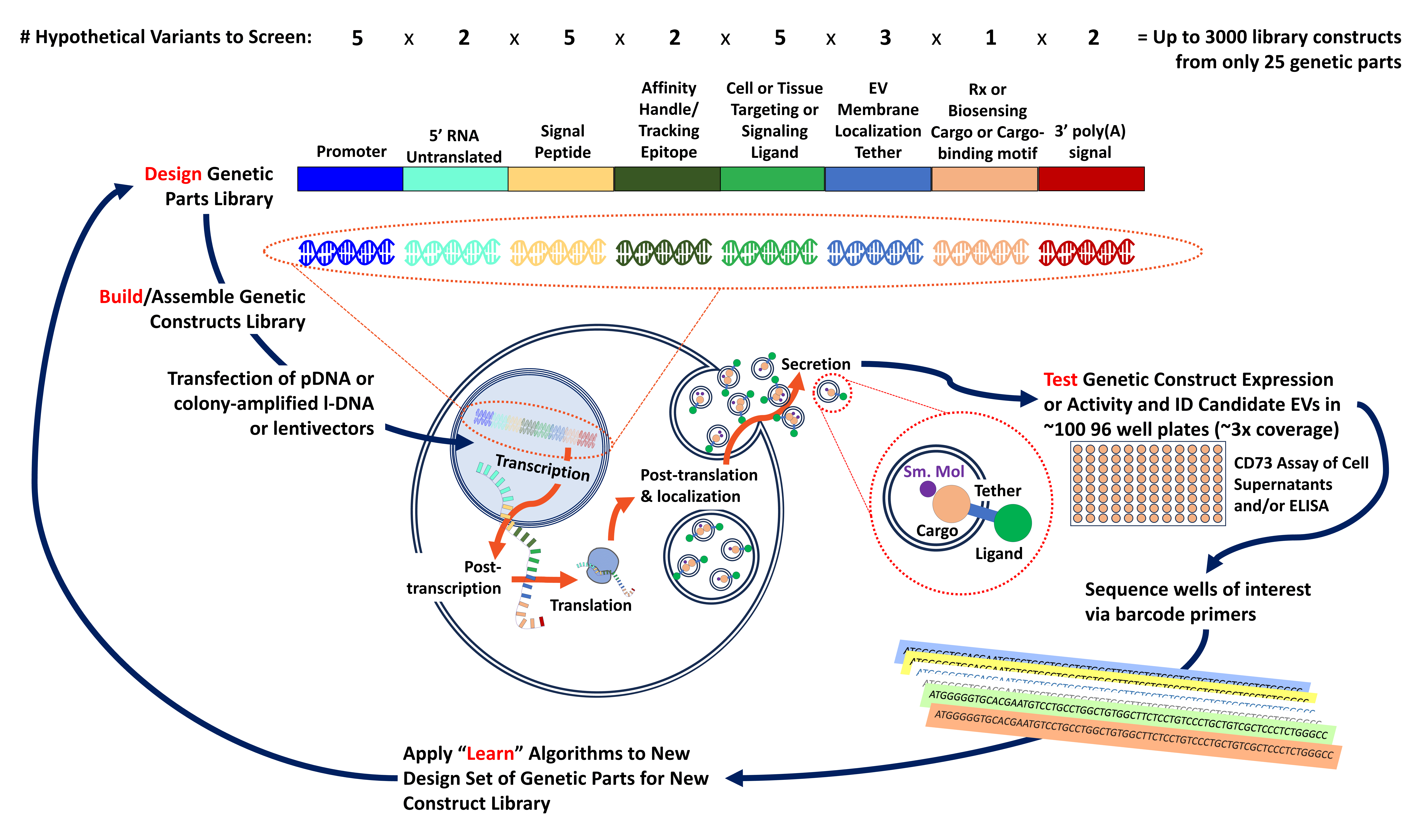
Figure 1. A hypothetical exosome/extracellular vesicle therapeutic candidate selection and optimization scheme that proceeds through an iterative synthetic biology engineering cycle of Design -> Build -> Test -> Learn.
Design: A virtual library of interchangeable component gene parts for bioproduction and biofunction of modified exosomes can be conceived by a gene designer informed by biological knowledge. 25 unique part variants are assigned 8 different classifications of a genetic part’s effector or controller biofunction (here). These 8 classifications span a range of variables to optimize artificial exosome expression and function at the level of transcription, post-transcription, translation, localization, tracking/tagging, cell targeting or ligand specificity, and effector/cargo biofunction.
Build: These gene parts are semi-randomly assembled into a material library of up to 3000 unique constructs, where each part fits into its ordinate component slot.
Test: Each unique construct is isolated from colony or plaque with no necessary a priori knowledge, then transfected at one construct per well (with ~3.2-fold library coverage) across 100 96-well plates alongside normalization controls (-/+), cell density originally plated to 8000 hMSCs per well. From ~9600 samples, obtain supernatant to ratio per-well data points via ELISA for presence of exosome artificial affinity epitope (e.g., myc-tag, Rituximab epitope, or AVITAG™) and assay to estimate total bioactive exosome activity (CD73 nucleotidase assay). Obtain PCRs from well addresses of interest via barcoded sequence IDs to unmask constructs that yield very high or very low levels or artificial exosomes.
Learn: Via the sequence-addressed subset of data, determine which assemblies of constituent gene parts yield exosomes with higher expression—while still permissive of a surrogate readout of exosome biofunction. Determine which parts or constructs need to be discarded from future mixes. Apply data to Design of Experiment (DOE) strategies toward subsequent iterations of the learning cycle until target product profile (TPP) criteria for an exosome candidate are met.
In Figure 1 (above), note the phases of “Design” and “Build.” Each category of genetic part (e.g., a DNA “biobrick” or “gblock,” etc.) relates to a distinct mechanism to exert control over the exosome’s cellular production or function—as if the individual cell were an instrument that executes on direct requests from your CMC team, its own bioreactor in microcosm. Each ordinate component slot of the synthetic gene program is directed to harmonize with host cellular machinery for optimized control of transcription, post-transcription, translation, post-translation and localization, and finally, secretion. There are also interchangeable effector parts within the protein-coding DNA sequence (cds), so there’s capability to swap in-and-out tissue targeting and/or multivalent signaling ligands, in vivo tracking and/or affinity purification steps, and intra-luminal therapeutic, diagnostic, or theragnostic cargoes.
Note what happens when you mix these example exosome modulator classes in a “grab bag,” full of LEGO®-like blocks that come together in a defined sequence but in different combinations. Even with very few variations per “block” (e.g., 1 to 5 per type; here totaling 25 discrete gene parts), there can quickly form thousands of permutations of gene constructs. In this visualized example, such combinations total up to 3000 variations. Some of the 3000 would certainly be expected to perform better than others for modified exosome/extracellular vesicle expression… some much worse. Yet what do these large numbers of samples mean for the “Test” phase of a synthetic biology loop?
A synbio-savvy exosome designer might almost certainly choose different variables and/or biofunction-assigned gene part slots to tailor for a larger or smaller experimental matrix. Yet for the sake of example, 3000 variable constructs in transfected cells means high-throughput screening (HTS). Does HTS sound intimidating to many smaller companies? Although such could seem a tall order, we’ll show how it may actually still be quite feasible to sift through this scheme multiple times in a few short months, even for a startup.
To maximize coverage of the whole library by a factor of 3.2, 3000 variants expands to ~9.6K gene non-sequence addressed constructs. These could be isolated in-house via bacterial colonies or phage plaques, then transfected as PCR-amplified linear DNAs. By hand, at 5 colonies per minute, e.g.
[a] dot colony with sterile pipet tip->
[b] dip tip into PCR reaction tube for linear DNA amplification->
and [c] inoculate/eject tip into 1 mL LB o/n culture for subsequent cryobanking or plasmid prep)
…computes to ~300 colonies per hour or ~33 hours of hands-on FTE labor, divided accordingly by time and persons (or automated colony picking instruments). This sure sounds like a lot of sweat (even with chill music jams in the earbuds). However, a good molecular candidate can easily save many more headaches and time later in bioprocess development! That is because favorable manufacturability parameters of the final product instance may already be “baked into” the earliest library variants to be mined from an adroitly run discovery program.
To save extra time, one might consider turning to entities such as Twist Bioscience or Ginkgo Bioworks, who are already highly adept at chip-based, automated DNA synthesis platforms to provide combinatorial libraries to use in applications like chimeric antigen receptors (CARs) or synthetic promoters for gene therapies. 9600 transfected cell assay sample points (including plate-by-plate normalization controls) translates to a “haystack” of ~100 96-well plates, a scale of operations to which 96-well electroporator hardware has long been applied. From these well supernatants, you could (i) capture approximate protein expression level of the sequence-addressed engineered exosome by ELISA, and (ii) capture a readout of overall extracellular vesicle bioactivity (CD73 activity) to use as in-sample normalization. Then, pick the “golden needles” from this haystack by sifting through the constructs that express both hi-synthetic epitope tag and hi-CD73, and prioritize “winners” with high tag-to-CD73 ratios. From sequence configurations and motifs that yield higher performance, you’ll next input these variants into a “Learn” algorithm toward design-of-experiment (DOE) calculations that facilitate subsequent round(s) of even better-tuned construct optimizations.
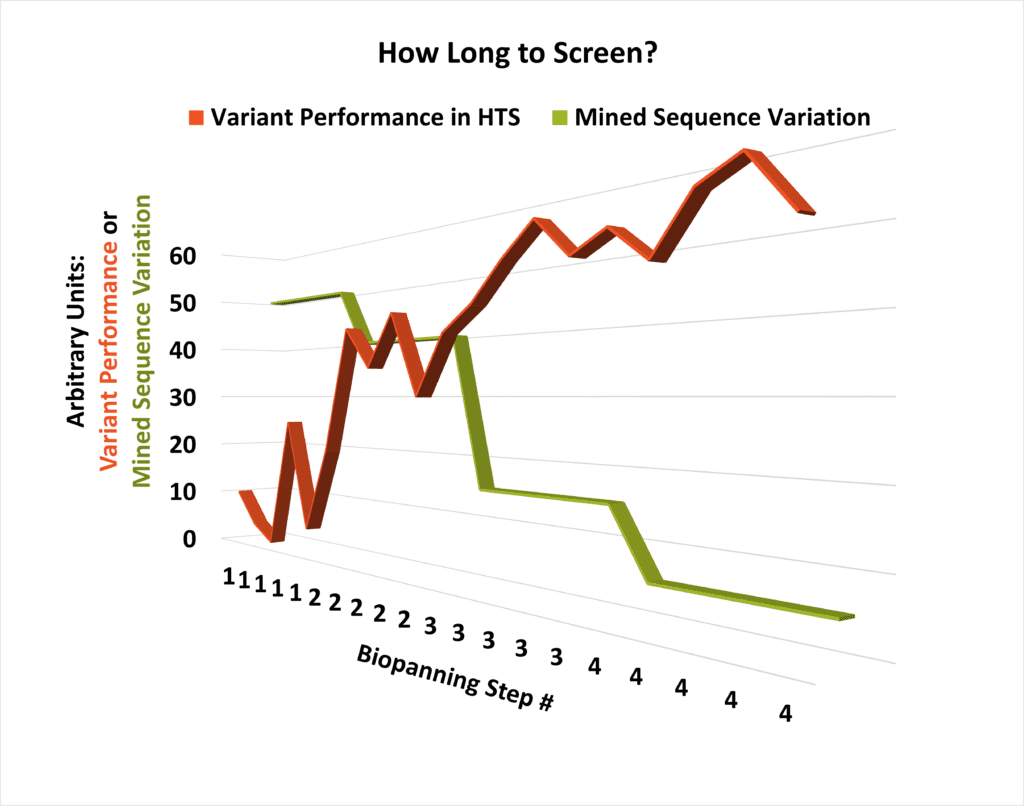
One critical question that arises during planning for multiple rounds of HTS is “How long will this take?” The answer isn’t instantly straightforward, but it can be arrived at empirically by comparing the bioassay performance of individual molecular candidates with the diversity of the library that produced them. As library variability begins to drop and performance starts plateauing at a high level, the number of necessary extra steps of panning the sequence space can be readily modeled (see conceptual schematic, left).
What is the best cell type to use for HTS testing of designer exosomes? Before you instinctually reach for your nearest flask of HEK-293 cells, recall that hMSCs remain the “go-to” cell type that many therapeutic exosome developers turn to. [19, 20, 21, 22, 23] Also note that, while easily transfectable, HEKs tend to adhere to the plastic much more loosely than hMSCs. Would repeated media washes between transfection and supernatant collection for extracellular vesicles tend to dislodge HEKs (and not hMSCs) in this kind of HTS assay? Maybe. Unlike HEK-293s, [24, 25] live hMSCs have a track record of in vivo safety across thousands of clinical trials over the last 20+ years, and these primary cells nominally exhibit a non-transformed phenotype. hMSC secretomes (which include extracellular vesicles and exosomes) can also recapitulate the therapeutic signals observed in vivo. While it’s no trouble to quickly fill an incubator with expanded HEKs, how many hMSCs would you need for an experiment with one hundred 96-well plates? If plated at 25K cells/cm2 for transfection the following day, this means 8000 cells per well, or a total of 80 million cells per HTS run. Turns out, hMSCs can handle this task most splendidly.
Unfortunately, most vendors of hMSCs only distribute in aliquots of 0.5 to 1 million cells per vial. However, RoosterBio readily provides hMSCs in 10–50 million or more cell aliquots at the lowest commercially available price on a per-million cell reckoning. RoosterBio’s high-performance media also enable predictable and rapid cell growth kinetics with no required mid-passage media exchanges. For example, a single vial of RoosterBio’s 10M-cell aliquot format, combined with RoosterNourish™ expansion medium, reliably expands out to 80M cells within 4-5 days after thawing. RoosterBio has developed SOPs for efficient transfection and/or transduction of hMSCs with the aid of media additives like RoosterGEM™ and is a leading bioprocessing expert in harvest of hMSC-conditioned media [26, 27] via specialized reagents such as RoosterCollect™-EV. It’s clear that companies like RoosterBio which catalyze solutions for “long” problems of process scale up are also equipped to help ameliorate “wide” problems of a different nature, i.e., scaling out for exosome/extracellular vesicle candidate lead discovery and optimization. Nevertheless, requirements for large cell numbers and liter-scale volumes of specialized media are quite similar for both process development and discovery arenas.
As Spock would propose, it’s “logical” to mind-meld the drug discovery engine with the CMC and process development, isn’t it? Harness the immense power of combinatorial molecular diversity on the back of a robust cell system that can breeze through the HTS, the bioreactor process, and the regulatory agencies. Even with a modality as complex as a genetically modified exosome/extracellular vesicle, we believe that it’s now far more feasible than ever to exploit existing synthetic biology, cell culture, and cell analytics resources to quickly file INDs for novel advanced therapeutics.
References
- Lenzini, S. Big Effects in Small Packages: What Are Extracellular Vesicles, Exosomes, & Microvesicles & Why Are They En Route to the Clinic? 2021; Available from: https://www.roosterbio.com/blog/big-effects-in-small-packages-what-are-extracellular-vesicles-exosomes-microvesicles-why-are-they-en-route-to-the-clinic/.
- Kalluri, R. and V. S. LeBleu, The biology, function, and biomedical applications of exosomes. Science, 2020. 367(6478). 10.1126/science.aau6977
- Stranford, Devin M, et al., Bioengineering multifunctional extracellular vesicles for targeted delivery of biologics to T cells. bioRxiv, 2022: p. 2022.05. 10.1101/2022.05.14.491879.
- Koh, E., et al., Exosome-SIRPalpha, a CD47 blockade increases cancer cell phagocytosis. Biomaterials, 2017. 121: p. 121-129. 10.1016/j.biomaterials.2017.01.004
- S, E. L. Andaloussi, et al., Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov, 2013. 12(5): p. 347-57. 10.1038/nrd3978
- Liu, Y., et al., Targeted exosome-mediated delivery of opioid receptor Mu siRNA for the treatment of morphine relapse. Sci Rep, 2015. 5: p. 17543. 10.1038/srep17543
- Yang, Y., et al., Virus-Mimetic Fusogenic Exosomes for Direct Delivery of Integral Membrane Proteins to Target Cell Membranes. Adv Mater, 2017. 29(13). 10.1002/adma.201605604
- Alvarez-Erviti, L., et al., Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol, 2011. 29(4): p. 341-5. 10.1038/nbt.1807
- Ridler, C., Designer exosomes alleviate neurotoxicity. Nat Rev Neurol, 2018. 14(6): p. 316-317. 10.1038/s41582-018-0006-y
- Kojima, R., et al., Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat Commun, 2018. 9(1): p. 1305. 10.1038/s41467-018-03733-8
- Haltom, A. R., et al., Engineered exosomes targeting MYC reverse the proneural-mesenchymal transition and extend survival of glioblastoma. Extracell Vesicle, 2022. 1. 10.1016/j.vesic.2022.100014
- Wan, T., et al., Exosome-mediated delivery of Cas9 ribonucleoprotein complexes for tissue-specific gene therapy of liver diseases. Sci Adv, 2022. 8(37): p. eabp9435. 10.1126/sciadv.abp9435
- Teng, F. and M. Fussenegger, Shedding Light on Extracellular Vesicle Biogenesis and Bioengineering. Adv Sci (Weinh), 2020. 8(1): p. 2003505. 10.1002/advs.202003505
- Kordelas, L., et al., MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia, 2014. 28(4): p. 970-3. 10.1038/leu.2014.41
- Maguire, C. A., et al., Microvesicle-associated AAV vector as a novel gene delivery system. Mol Ther, 2012. 20(5): p. 960-71. 10.1038/mt.2011.303
- Meliani, A., et al., Enhanced liver gene transfer and evasion of preexisting humoral immunity with exosome-enveloped AAV vectors. Blood Adv, 2017. 1(23): p. 2019-2031. 10.1182/bloodadvances.2017010181
- Breuer, C. B., et al., In vivo engineering of lymphocytes after systemic exosome-associated AAV delivery. Sci Rep, 2020. 10(1): p. 4544. 10.1038/s41598-020-61518-w
- Cheng, M., et al., Neutralizing Antibody Evasion and Transduction with Purified Extracellular Vesicle-Enveloped Adeno-Associated Virus Vectors. Hum Gene Ther, 2021. 32(23-24): p. 1457-1470. 10.1089/hum.2021.122
- Caplan, A. I. and D. Correa, The MSC: an injury drugstore. Cell Stem Cell, 2011. 9(1): p. 11-5. 10.1016/j.stem.2011.06.008
- Caplan, A. I., MSCs: The Sentinel and Safe-Guards of Injury. J Cell Physiol, 2016. 231(7): p. 1413-6. 10.1002/jcp.25255
- celltrials.org. available from Cell Trials Data. 2022; Available from: https://celltrials.org/public-cells-data/msc-trials-2011-2020/.
- Pittenger, M. F., et al., Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen Med, 2019. 4: p. 22. 10.1038/s41536-019-0083-6
- Hildreth, C. . MSC Therapies: Globally Approved Mesenchymal Stem Cell Therapeutics. BioInformant 2019; Available from: https://bioinformant.com/msc-therapies/.
- Shen, C., et al., The tumorigenicity diversification in human embryonic kidney 293 cell line cultured in vitro. Biologicals, 2008. 36(4): p. 263-8. 10.1016/j.biologicals.2008.02.002
- Lin, Y. C., et al., Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat Commun, 2014. 5: p. 4767. 10.1038/ncomms5767
- Cruz, C., Haylock, D. Manufacturing Advances Unlock the Therapeutic Potential of MSC-Derived Exosomes. 2023; Available from: https://tinyurl.com/roosterwebinar202306.
- Lenzini, Stephen. Extracellular Vesicle/Exosome Upstream Process Development: Maximizing Productivity to Accelerate Clinical Adoption. RoosterBio Blog 2022; Available from: https://www.roosterbio.com/blog/extracellular-vesicle-exosome-upstream-process-development-maximizing-productivity-to-accelerate-clinical-adoption/.